Материалы представлены из учебного пособия РУДН
Анемии. Клиника, диагностика и лечение / Стуклов Н.И., Альпидовский В.К., Огурцов П.П. – М.: ООО «Медицинское информационное агентство», 2013. – 264 с.
Копирование и тиражирование материалов без указания авторов запрещено и преследуется по закону.
Талассемии – это группа наследственных заболеваний, относящихся к количественным гемоглобинопатиям, при которых имеет место нарушение синтеза полипептидных цепей глобина β или α, что приводит к уменьшению образования одной или нескольких фракций нормального гемоглобина или, в редких случаях, – формированию гемоглобина, не встречающегося у здоровых людей.
При гомозиготных состояниях этот дефект синтеза цепей глобина проявляется гемолитической анемией разной степени выраженности. При гетерозиготных β-талассемиях, когда патологический ген наследуется от одного из родителей, а также у некоторых больных с гомозиготной α-талассемией заболевание проявляется слабовыраженной гипохромной анемией, а признаки гемолиза отсутствуют.
Впервые талассемия была описана Cooley и Lee в 1925 году при наблюдении 5 детей из итальянской семьи. У всех детей в семье наблюдалась гипохромная гемолитическая анемия со значительным увеличением селезенки и печени, изменения костей скелета. Заболевание назвали болезнью Кули. Позднее были описаны более легкие формы, и в 1936 году болезнь получила название «талассемия».
В зависимости от нарушения синтеза α- или β-цепей глобина выделяют две основные формы заболевания: α- и β-талассемию.
β-талассемии
β-талассемия обусловлена рядом мутаций в локусе β-глобина на 11 хромосоме, нарушающих синтез β-цепей. Эти мутации могут происходить на разных стадиях экспрессии гена, отвечающего за синтез β-цепей: дефект ДНК, нарушение гена-регулятора цепей глобина или транспортной РНК. При ограничении транскрипции т-РНК обычно снижается синтез β-цепей – β + -талассемия. При β 0 -талассемии мутация происходит в зоне кодирования и вызывает полную остановку синтеза β-цепей. Всего описано более 100 различных мутаций локуса β-глобина, которые передаются доминантно, но иногда возникают в результате спонтанных мутаций.
Патогенез клинических проявлений при гомозиготной β-талассемии связан не только с недостаточной продукцией нормального гемоглобина, но и с относительной избыточной выработкой малорастворимых α-цепей, свободные α-цепи образуют тетрамеры. В количественном отношении α-цепи вырабатываются нормально. Эти избыточные α-цепи в виде внутриклеточных преципитатов выпадают в осадок, разрушая мембрану клеток-предшественников эритроцитов, вызывая их внутрикостномозговую гибель (неэффективный эритропоэз) и гемолиз уже образовавшихся эритроцитов. Все это приводит к развитию основного симптома гомозиготной талассемии – анемии.
В ответ на развитие тканевой гипоксии, сопровождающей анемию, почки значительно повышают выработку эритропоэтина, который стимулирует формирование кроветворной ткани в костях, где она обычно отсутствует, вызывая деформацию костей (башенный череп, монголоидность и др.), а также образование экстрамедуллярного кроветворения в печени и селезенке с их выраженным увеличением.
β-талассемия широко распространена в странах Средиземного моря (Италия, Греция, Кипр, Турция и др.), Центральной и Восточной Африке, Ближнем и Среднем Востоке, Индии, Юго-Восточной Африке. Широкая распространенность заболевания, предполагается, является результатом лучшей переносимости малярии больными-гетерозиготами по β-талассемии – механизм остается неясным.
Выраженность клинических проявлений при гомозиготной β -талассемии зависит от степени нарушения синтеза β-цепей.
Самая тяжелая форма, или болезнь Кули, обозначается как большая β 0 -талассемия. При этой форме полностью отсутствует синтез β-цепей.
Анемия при большой талассемии развивается уже к первому году жизни ребенка и сопровождается отставанием в психическом и физическом развитии. Со временем анемия прогрессирует, появляется желтушность кожи и склер, значительно увеличиваются селезенка и печень, нередко на коже ног образуются трофические язвы. Характерен внешний вид больных детей с болезнью Кули: непропорционально большая деформированная голова («башенный череп»), приплюснутый нос, суженые глазные щели и выпуклые скулы (признаки «монголоидности»). Дети резко отстают в развитии, отмечается повышенная восприимчивость к инфекциям, половое недоразвитие.
При рентгенологическом исследовании трубчатых костей отмечается истончение кортикального слоя и расширение костномозгового канала. Для рентгенографической картины костей черепа характерно расширение диплоического пространства костей свода черепа и появление радиальной исчерченности – вид «ёжика» или «щетки». Сходные изменения структуры наблюдаются в метафизах трубчатых костей, плоских костях таза, лопатках и ребрах.
В анализах крови – глубокая анемия, концентрация гемоглобина – 30-50 г/л, выраженная гипохромия, анизоцитоз, значительная мишеневидность эритроцитов, базофильная пунктация. При электрофорезе гемоглобина эритроцитов 98% составляет HbF , 2-3% – HbA 2 и большое количество свободных α-цепей.
Ранее прогноз при этой форме заболевания был крайне неблагоприятным, поскольку большинство детей погибало в возрасте 3-4 лет. В настоящее время выздоровление детей с той тяжелой патологией возможно после проведения трансплантации аллогенного костного мозга.
При среднетяжелой и промежуточной форме β + -талассемии смягчение тяжести заболевания обусловлено частичным сохранением синтеза β-цепей, а, следовательно, присутствием в эритроцитах нормального HbA .
От способности синтезировать β-цепи зависит тяжесть течения заболевания. При незначительной продукции β-цепей эта форма β + -талассемии, тип I , обозначается как среднетяжелая и по клинике приближается к болезни Кули. Больные доживают до школьного возраста, а в их эритроцитах преобладает HbF 60 — 80%, HbA 2 3-9%, свободные α-цепи и появляется небольшое количество HbA .
Больные с β + -талассемией, тип II , с более выраженной продукцией β-цепей, описывается как промежуточная форма ( thalassemiaintermedia ), с менее выраженной анемией. Больные доживают до зрелого возраста, при адекватной гемотрансфузионной терапии практически не отстают в развитии и способны заканчивать школы, колледжи и даже университеты.
Диагностика гомозиготной β-талассемии не вызывает больших трудностей:
— признаки гемолитической анемии с детства;
— отставание в развитии и деформация скелета (болезнь Кули и среднетяжелая форма);
— мишеневидность и гипохромия эритроцитов;
— преобладание при электрофорезе гемоглобина HbF и свободных α-цепей, увеличение А2 Hb ;
— молекулярная диагностика (исследование мутаций генов, кодирующих цепи Hb ).
Большим достижением в лечении тяжелой и среднетяжелой форм β-талассемии стало применение аллогенной трансплантации костного мозга (чаще от брата или сестры), позволившее в 80 – 90% добиться выздоровления ранее некурабельных больных.
Больные с промежуточной формой β + -талассемии, как и больные с тяжелой формой заболевания, не могут жить без периодических трансфузий эритроцитарной массы, частота которых регулируется уровнем концентрации гемоглобина.
Для нормального развития ребенка рекомендуется поддерживать концентрацию гемоглобина на уровне 100 – 110 г/л. Регулярные гемотрансфузии, в сочетании с повышенным всасыванием железа в кишечнике у анемизированных больных создает опасность развития гемосидероза с поражением сердца (кардиомегалия), печени (фиброз), поджелудочной железы (сахарный диабет), почек (почечная недостаточность) и других органов. Поэтому гемотрансфузионная терапия должна сочетаться с применением хелатирующих лекарственных препаратов (десферал, эксиджад), которые способны связывать свободное сывороточное железо, внутриклеточное железо из гепатоцитов, извлекать его из комплекса с трансферрином и ферритином.
Десферал вводится внутривенно, длительно, по 1-2 г препарата на каждые 500 мл донорской эритроцитарной массы. Возможно длительное подкожное введение с помощью инжектора с одновременным определением сывороточного ферритина.
В последние годы разработан препарат со свойствами десферала – эксиджад (деферазирокс), обладающий выраженным преимуществом, поскольку он может применяться внутрь, по 20 мг/кг массы тела больного.
При массивной спленомегалии рекомендуется проведение спленэктомии. Показания:
— прогрессирующе увеличение размеров селезенки (>8 см из-под края реберной дуги),
— увеличение потребности в гемотрансфузиях более, чем на 50% от исходного уровня в течение 6 месяцев,
— при повышении потребности в эритроцитарной массе более, чем на 250 мл в течение года.
α-талассемии
α-талассемия впервые была описана в 1955 году. Выявляется в Греции, Таиланде, Нигерии, Азербайджане, Дагестане. В основе α-талассемии лежит нарушение синтеза α-цепей глобина. Поскольку эти цепи входят в состав всех нормальных фракций гемоглобина, при α-талассемии происходит равномерное снижение их синтеза.
Две почти идентичные копии гена α-глобина находятся на хромосоме 16. Наиболее часто встречается потеря одного или нескольких их этих 4 генов – 80-85% случаев α-талассемии.
Клинические проявления α-талассемии напрямую коррелируют со степенью нарушения синтеза α-глобиновых цепей. При данном виде аномалии симптомы менее выражены, чем при β-талассемии, что связано с наличием 4 генов α-глобина. Адекватное количество α-цепей образуется до тех пор, пока не утрачиваются 3 или 4 гена. Кроме того, β4-тетрамер (или HbH ), образующийся при недостатке α-цепей, более растворим, чем тетрамер α4. Вследствие этого даже при выраженном нарушении синтеза α-цепей при α-талассемии гемолиз гораздо менее выражен, а эритропоэз более эффективен, чем при β-талассемии.
Гемоглобин Барт – гомозиготная α-талассемия I – поражены все 4 гена, ответственные за синтез α-цепей – самая тяжелая форма α-талассемии. Встречается такая форма только в странах Юго-Восточной Азии. Это состояние не совместимо с жизнью. Беременность в подобных случаях заканчивается самопроизвольным выкидышем, мертворождением или гибелью плода в первые часы жизни (водянка плода), так как у плода не синтезируется фетальный гемоглобин ( α 2 γ 2 ). Свободные γ-цепи образуют тетрамеры (γ4), так называемый гемоглобин Барт. Такой гемоглобин обладает высоким сродством к кислороду, что приводит к гипоксии тканей плода, сердечной недостаточности, и, в итоге, к гибели плода. В крови – выраженные эритробластемия, гипохромия, макроцитоз, мишеневидность, анизо- и пойкилоцитоз. Электрофоретически в эритроцитах обнаруживается Hb Барт (80-90%) в сочетании с HbH .
H -гемоглобинопатия
Н-гемоглобинопатия вызвана утратой или дисфункцией трех α-глобиновых генов. Гемоглобин Н представляет собой тетрамер β4, образующийся при избытке β-цепей. Клинически близок к промежуточной форме β-талассемии.
Заболевание проявляется к концу первого года жизни гемолитической анемией различной степени выраженности. Характеризуется нетяжелым течением, значительным увеличением селезенки и печени. Отмечается слабо выраженная желтуха из-за увеличения непрямого билирубина. Концентрация гемоглобина – в пределах 70 – 80 г/л, выявляется гипохромия, мишеневидность и базофильная пунктация эритроцитов.
На фоне приема лекарственных препаратов или интеркуррентной инфекции могут развиваться гемолитические кризы с падением уровня гемоглобина до 50 г/л. При электрофорезе доля HbH составляет 5-30% при нормальном уровне HbF и снижении HbA 2 .
Лечение практически не отличается от лечения гетерозиготной β-талассемии.
источник
Сидеробластная анемия
Анемия хронического заболевания
Определение.Нарушение утилизации запасов железа в ретикулоэндотелиальной системе. Может быть как микроцитарной, так и нормоцитарной.
Этиология.Анемия может сопровождать практически любое хроническое воспалительное, инфекционное или онкологическое заболевание.
Клинические проявления.Тяжесть симптомов зависит от степени выраженности анемии, как было описано выше. Сопустствующие специфические проявления зависят от основного заболевания.
Диагностика.Уровень ферритина плазмы нормальный или повышенный. Сывороточное железо и общая железо-связывающая способность плазмы понижены. Число ретикулоцитов низкое.
Лечение.Терапия основного заболевания. Препараты железа и эритропоэтин бесполезны, за исключением почечной этиологии анемии.
Определение.Микроцитарная анемия, вызванная нарушением синтеза гемоглобина вследствие задержки железа в митохондриях ядросодержащих эритроцитов.
Этиология.Существуют как наследственные так и приобретенные формы. В основе наследственной формы лежит дефект аминолевулоновой синтетазы или аномалии метаболизма витамина В6. Приобретенную форму вызывают такие препараты как хлорамфеникол, изониазад или алкоголь. Также к сидеробластной анемии может привести отравление железом. Имеется связь с миелодиспластическими синдромами и рефрактерной анемией. У небольшого числа пациентов заболевание перерастает в острый миелогенный лейкоз.
Клинические проявления.Тяжесть симптомов зависит от степени выраженности анемии, как было описано выше. Не существует симптомов, достаточно специфических, чтобы поставить диагноз сидеробластной анемии без подробных данных лабораторных методов исследования.
Диагностика.Уровень ферритина плазмы в норме. Насыщение трансферрина очень высокое, поэтому общая железо-связывающая способность плазмы очень низкая. Железо сыворотки высокое. Наиболее специфическим тестом является окраска эритроцитов костного мозга прусским голубым для выявления кольцевидных сидеробластов.
Лечение.Отмена провоцирующего препарата. Лечение отравления свинцом. У некоторых пациентов с наследственной формой эффективной оказывается терапия пиридоксином.
Определение.Генетически обусловленное нарушение синтеза альфа или бета цепи глобина в молекуле гемоглобина, что приводит к гипохромной, микроцитарной анемии.
Этиология.Делеция гена приводит к развитию заболевания разной степени тяжести. Альфа-цепь гемоглобина кодируют четыре гена. Может иметь место делеция одного, двух, трех или всех четырех генов. Бета-талассемия развивается при мутации одного или двух генов. Альфа-талассемия более распространена среди Азиатов. Бета-талассемия чаще поражает жителей Средиземноморского региона.
Клинические проявления.Тяжесть заболевания зависит от количества аномальных генов. В случае альфа-талассемии отсутствие одного гена никак не проявляется. Все показатели крови в норме. При делеции двух генов развивается легкая анемия, гематокрит на уровне 30 – 40 %, СОК очень низкий. Поражение трех генов приводит к выраженной анемии с гематокритом 22 – 32 % и очень низким СОК. Бета-талассемия проявляется легкой анемией с выраженным микроцитозом (низкий СОК). Больные с отсутствием четырех генов погибают внутриутробно вследствие образования тетрад гамма-цепей, называемых гемоглобином Бартса. Невозможно отличить анемию, вызванную малыми талассемиями, неважно альфа или бета, от ЖДА или анемий вследствие других причин, вроде дефицита фолиевой кислоты, только на основании клинических проявлений. Проявления не зависят от этиологии. Проявления зависят от степени тяжести анемии. Пациенты с большой бета-талассемией гомозиготны по генам, кодирующим синтез бета-цепей гемоглобина. У этих пациентов с большой бета-талассемией, также известной как анемия Кули, развивается тяжелая анемия через 6 месяцев после рождения, когда обычно происходит смена фетального гемоглобина на взрослый. Проявления очень выраженные, включают задержку развития, гепатоспленомегалию, желтуху, деформации костей вследствие экстрамедуллярного гематопоэза. Позднее развивается гемохроматоз, цирроз и застойная сердечная недостаточность из-за хронической анемии и постоянных переливаний крови.
Диагностика.На наличие малой формы талассемии указывает легкая анемия с выраженным микроцитозом. При большой бета-талассемии наблюдаются тяжелые проявления: увеличение селезенки, аномалии костей, описанные выше. Признаком обеих форм талассемии является микроцитарная анемия при нормальных результатах исследования железа крови. Точно тип талассемии определяют с помощью электрофореза гемоглобина. При бета-талассемии повышаются уровни гемоглобина F и гемоглобина А2. У больных с альфа-талассемией уровни этих видов гемоглобина будут в норме. Тетрады бета-цепей называются гемоглобин Н. Он появляется при альфа-талассемии в случае отсутствия трех из четырех генов. Мишеневидные эритроциты определяются при всех формах талассемий. Разброс ширины эритроцита (РШЭ) в норме при всех формах, так как все эритроциты имеют одинаковый размер.
Лечение.Малые формы как бета так и альфа талассемии не требуют лечения. При большой бета-талассемии требуются переливания крови один или два раза в месяц. Постоянные переливания приводят к перегрузке железом, что в свою очередь требует лечения дефероксамином. Спленэктомия устраняет основной очаг разрушения эритроцитов, что снижает потребность в переливаниях эритроцитарной массы. У небольшого числа больных возможно выполнение пересадки костного мозга.
Дата добавления: 2014-12-16 ; Просмотров: 857 ; Нарушение авторских прав? ;
Нам важно ваше мнение! Был ли полезен опубликованный материал? Да | Нет
источник
При тяжелой талассемии анемия неизменно тяжелая и резко гипохромная, при этом значения гемоглобина колеблятся от 5 до 7 г/100 мл крови, а показатели гематокрита и численности красных кровяных клеток соответственно уменьшаются. Постоянные эритроцита НЕМ и СНЕМ меньше нормы, соответственно недостаточности гемоглобина в эритроцитах. Вместе с тем, показатель РОЭ колеблется в соответствии с недостаточностью фолиевой кислоты и связанным с ней вторичным макроцитозом.
Мазок периферической крови характерен и с самого же начала исследования подсказывает диагноз. Гипохромия резко выражена, ей сопутствует анизопойкилоцитоз высокой степени. Многочисленные точечные и частые капельные гематии и овалоциты, наряду с большим числом красных кровяных телец искаженного очертания и шизоцитов различной формы и величины, в целом характеризуют мазок при анемии Кули.
Звездочкообразные гематии с подвернутыми краями напоминают «мексиканскую шляпу», при этом иногда они макромегалоцитные (12—15 u). В случаях очень низкого показателя гемоглобина отдельные гематии как бы лишены содержания, в связи с чем принимают аспект «эритроцитной тени». Часто отмечается наличие красных кровяных телец с кольцами Cabot, телец Jolly и базофильными точками. Неизменно присутствие эритробластов (5—20/100 лейкоцитов), при этом, после удаления селезенки, их численность в несколько раз больше лейкоцитов (300—500/100 лейкоцитов).
Выраженная полихроматофилия мазка отражает повышенный ретикулоцитоз (5—15%).
При легкой талассемии анемия умеренная (гемоглобин от 7 до 10 г/100 мл) или небольшая, причем в формах отягчения могут наблюдаться нормальные значения гемоглобина. Степень эритроцитной беспорядочности значительно меньше, чем при тяжелой талассемии. Мазок периферической крови отражает общий аспект микроцитной гипохромной анемии. Тем не менее пойкилоцитоз, наличие звездочкообразных и точечнобазофильных красных кровяных телец делает возможным отличить это заболевание от однородной микроцитно-гипохромной картины железодефицитной анемии.
Небольшая полихроматофилия соответствует слегка увеличенной численности ретикулоцитов (2—5%). Эритробласты редкие (1—2 на несколько сот лейкоцитов) или, как это бывает в большинстве случаев, просто отсутствуют.
Умеренный лейкоцитоз постоянное явление при тяжелой талассемии (10 000—20 000/мм3), ему нередко сопутствует отклонение влево лейкоцитной формулы. У большинства больных наблюдается и небольшой рост показателя численности тромбоцитов. В принципе, при легких формах, число лейкоцитов и тромбоцитов укладывается в норму или у высшего предела.
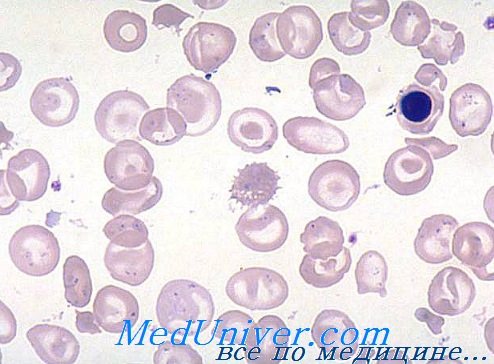 Мазок при бета-талассемии
Мазок при бета-талассемии
Косвенный билирубин увеличен (1—3 мг/100 мл), показатели уробилиногена в моче и выделения стеркобилиногена с испражнениями — весьма высокие. При легкой талассемии все эти признаки разрушения красных кровяных клеток существуют, но в значительно сокращенном виде в зависимости от клинической формы и степени расплавления крови.
Осмотическая устойчивость эритроцитов в гипотонном солевом растворе почти неизменно повышена, общее расплавление крови часто отмечается при концентрации примерно 0,2 г NaCl/100 мл или даже меньше в случае выраженного платицитоза. Для диагностирования талассемии реакция весьма полезна, но отнюдь не патогномонична. Отмечаются случаи железодефицитиой анемии с высокой осмотической устойчивостью, подобно случаям талассемии-отягчение, при которой показатели близки к норме.
Сидервмия, общая способность связывания сидерофилина (СС), латентная способность (ЛС), коэффициент насыщения (КН) сидерофилина железом выявляют степень нагрузки организма железом. В большинстве случаев тяжелой талассемии СС меньше нормы (300—330 мкг), показатель сидеремии всегда повышен и КН колеблется от 60 до 100%, подобно тому как наблюдается при гемохроматозе. Окраска на определение костномозгового гемосидерина выявляет увеличенное количество последнего в макрофагах и значительный процент сидеробластов.
При легкой талассемии сидеремия несколько повышена или нормальна, причем в отдельных случаях показатель СС несколько занижен а значения КН сидерофилина укладываются в перделы от 33 до 60%. Вообще процент сидеробластов превышает норму. Определение сидеремии, СС, КН и костномозгового железа особо полезны при дифференциальной диагностике с гипохромной железодефицитиой анемией.
В условиях тяжелой талассемии костный мозг отличается выраженной гиперплазией с явным преобладанием эритробластического ряда (Г/Э = 1/4—1/8). В случае недостатка фолиевой кислоты некоторые эритробласты приобретают мегалобластовидный аспект. При легкой талассемии костномозговой мазок выявляет наличие обильной клеточной массы с умеренным преобладанием эритробластического ряда. В принципе исследование костного мозта не обязательно для постановки диагноза, тем не менее оно полезно для определения запасов железа, в основном при легкой талассемии с электрофорезом гемоглобина в пределях нормы или при осложнениях с хронической кровоточивостью и недостатком железа.
Электрофорез гемоглобина основной способ для уточнения диагноза бета-талассемии. Тяжелая талассемия или анемия Кули характеризуется весьма высоким показателем гемоглобина F (N > 2% у детей старше 1 года), составляющим 20—90% всего гемоглобина, в остальной части отмечается наличие гемоглобин А и нормальные или завышенные значения гемоглобин А2. Наиболее часто значения гемоглобин F составляют 30—60%. В случаях гомозиготной бета-талассемии вида дельта-бета все количество гемоглобина состоит лишь из вида F, при полном отсутствии видов А и А2. Значения Гб в результате электрофореза следует сопоставить со значениями химического теста на определение щелочноустойчивого гемоглобина, в целях исключения аномального гемоглобина, электрофоретическая миграция которого может оказаться одинаковой с гемоглобином F.
Гетерозиготная бета-талассемия характеризуется ростом показателя гемоглобина А2 и/или гемоглобина F, в процентном выражении. Большинство случаев гетерозиготной талассемии (примерно 90%) относятся к виду I при этом показатель гемоглобина А2 колеблется в пределах от 4 до 6% (N > 3%). Значения от 3 до 4%, в частности от 3 до 3,5% следует толковать осторожно и сопоставлять с результатами остальных исследований. Примерно в 50% случаев гетерозиготной бета-талассемии, с высоким показателем гемоглобина А2, отмечается и слегка повышенный показатель гемоглобина F (2—5%), причем для их определения применяется тест на устойчивость к щелочам.
Бывают случаи легкой бета-талассемии, осложняющиеся недостатком железа (повторные роды, хроническая кровоточивость и пр.), при которых значения гемоглобина А2 при электрофоретическом исследовании укладываются в норму. После проведения железотерапии повторный электрофорез выявляет рост показателя гемоглобина А2 за пределы нормы.
Случаи гетерозиготной бета-талассемии, характеризующиеся ростом лишь гемоглобина F редки (среди нашего населения примерно 10% всех гетерозиготов), при этом соответствующий показатель составляет примерно 2—30%, наиболее часто 5—15%; у подобных больных гемоглобин А2 нормальный или заниженный, остальную долю — составляет гемоглобин А. Такие случаи следует отличать от физиологических или патологических состояний с высоким показателем гемоглобин F, таких как, беременность, бирмеровская и сидероакрестическая анемия, лейкемия и пр.
Диагностирование бета-талассемии относительно несложное. Проявление болезни в детстве, клинические и гематологические признаки тяжелой гемолитической анемии подсказывают диагноз наследственной гемолитической анемии. Аспект мазка периферической крови исключает остальные виды наследственной гемолитической анемии, в первую очередь врожденного сфероцитоза. Электрофорез и определение щелочноустойчивого гемоглобина выявляют весьма высокий показатель гемоглобина F, и тем самым подтверждают диагноз.
При легкой форме бета-талассемии клиническое и гематологическое исследования менее убедительны. Наиболее частая ошибка заключается в определении заболевания как гипохромная железодефицитная анемия. Однако пойкилоцитоз, звездочкообразные красные кровяные тельца, базофильноточечные гематии, равно как и признаки расплавления крови свидетельствуют о наличии легкой формы талассемии.
Высокие показатели сидеремии и костномозгового гемосидерина лишний раз подтверждают это предположение. Диагноз подтверждается электрофорезом и определением щелочноустойчивого гемоглобина, поскольку эти исследования выявляют высокие показатели гемоглобина А2 и/или гемоглобина F. Исследования в семейном плане подтверждают наследственный характер заболевания. Так, при тяжелой талассемии, как у отца, так и у матери выявляется наличие легкой талассемии, в то время как при легкой форме этого заболевания, по меньшей мере у одного из них.
источник
Наследственные заболевания встречаются довольно часто в нашей жизни: одни из них не причиняют сильного вреда для организма, другие же могут привести к летальному исходу. Одним из заболеваний, передающихся по наследство, является талассемия. Попытаемся разобраться, кому она может передаться, как отразиться на человеке и можно ли с этим жить.
Талассемия – заболевание, в основе которого лежит нарушение синтеза цепей гемоглобина. Она относится к количественным гемоглобинопатиям.
Причиной талассемии являются точечные мутации или делеции в генах, кодирующих цепи гемоглобина. В результате это может привести к уменьшению синтеза или полному отсутствию одной из цепей в организме. Другая цепь образует неадекватные тетрамеры гемоглобина, что приводит к разрушению эритроцитов и гемолитической анемии.
Гемоглобин – белок содержащийся в эритроцитах, отвечающий за перенос кислорода к тканям и углекислого газа от них.
Гемоглобин (речь идёт о HbA) состоит из четырёх цепей: двух альфа-субъединиц и двух бета-субъединиц. Такой гемоглобин составляет 97% от общего содержания его в эритроцитах.
Каждая из цепей гемоглобина связывается с его небелковой частью – гемом.
Так вот при талассемии нарушается синтез одной из цепей гемоглобина: либо альфа, либо бета. По этому принципу имеется классификация талассемии на:
По степени тяжести выделяют талассемию:
- лёгкой степени;
- средней степени;
- тяжёлой степени.

Талассемии — это гетерогенная группа наследственно обусловленных гипохромиых анемий, имеющих различную тяжесть течения, в основе которых лежит нарушение структуры цепей глобина. Многочисленные типы талассемии с разнообразными клиническими и биохимическими проявлениями связаны с дефектом в любой из полипептидных цепей (α, β, γ, 5). В отличие от гемоглобинопатии при талассемиях отсутствуют нарушения в химической структуре гемоглобина, но имеется искажение количественных соотношений НbА и HbF. При некоторых типах α-талассемий встречаются тетрамерные формы, такие как HbH (β=4), Hb Bart’s (γ=4). Синтез полипептидных цепей может полностью отсутствовать, что отмечается при β 0 типе β-талассемии, или характеризоваться частичной недостаточностью (β + тип).
Молекулярный патогенез синдрома талассемии выражается в избыточном синтезе α- или β-цепей гемоглобина. Установлена корреляция между избытком синтезируемых α-цепей при β-талассемии и выживаемостью эритроидных клеток. Пониженная выживаемость эритроидных клеток с избыточным синтезом α-цепей при большой форме β-талассемии приводит к неэффективному эритропоэзу.
Доказано, что изолированные цепи гемоглобина, особенно α-цепи, более лабильны и менее устойчивы к денатурирующим воздействиям по сравнению с тетрамером гемоглобина. Их окисление и последующая агрегация вызывают повреждение мембраны. Этот процесс сопровождается перекисным окислением липидов и белков мембраны эритроцитов высокоактивными свободными радикалами кислорода, образующимися при самоокислении изолированных цепей. Оба процесса вызывают гибель эритроидной клетки.
Синдром водянки плода с гемоглобином Барта — это наиболее тяжелая форма α-талассемии, обусловленная гомозиготной α-талассемией-1 (поражены все четыре гена, по два на каждой хромосоме) и, таким образом, не продуцируется функциональный гемоглобин, за исключением эмбриональной стадии, на которой синтезируются α-подобные цепи. Свободный β-глобин образует тетрамеры, называемые Нb Барта, обладающие очень высоким сродством к кислороду. Нb Барта не высвобождает гемоглобин в ткани плода, из-за чего возникают тканевая асфиксия, отек, застойная сердечная недостаточность и наблюдается клиническая картина водянки плода.
Гемоглобин Барта встречается почти исключительно у жителей Юго-Восточной Азии, у которых наблюдается преимущественно цис-делеция α-глобиновых генов. Дети с водянкой плода нежизнеспособны, смерть наступает либо in utero, либо в первые часы жизни. При осмотре плод бледный, отечный, с массивной и рыхлой плацентой. Видны петехии на коже. Имеются дефекты развития легких. Сердце увеличено, выражена гипертрофия обоих желудочков. Вилочковая железа увеличена. Увеличение печени более выражено, чем селезенки. Определяются асцит, выпот в плевре и перикарде, часто неопущение яичек и гиноспадия. В различных тканях и органах обнаруживаются отложения гемосидерина вследствие тяжелого гемолиза у плода. Уровень общего гемоглобина составляет 30-100 г/л. Состав гемоглобина: гемоглобин Барта 70-100 % со следами гемоглобина Portland; НbН, HbA, HbA2, HbF отсутствуют. В крови резкая эритробластемия с гипохромными макроцитами, мишеневидные клетки, выраженные анизо- и пойкилоцитоз.
 [1], [2], [3], [4], [5]
[1], [2], [3], [4], [5]
Бета-талассемия (β-тал) — гетерогенная группа заболеваний, характеризующаяся уменьшением или отсутствием синтеза β-глобиновых цепей. В зависимости от тяжести состояния выделяют 3 формы β-талассемии: большую, промежуточную и малую. Тяжесть клинических проявлений прямо пропорциональна степени дисбаланса глобиновых цепей.
[6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16]
Альфа-талассемия — это группа заболеваний, распространенных в Юго-Восточной Азии, Китае, Африке, Средиземноморье. Две почти идентичные копии гена α-глобина находятся на хромосоме 16. В 80 — 85 % случаев α-талассемии происходит потеря одного или нескольких из этих четырех генов. У остальных больных эти гены сохраняются, но не функционируют.
Клинические проявления α-талассемии коррелируют со степенью нарушения синтеза α-глобиновой цепи, однако они обычно выражены слабее, чем при β-талассемии. Это связано, во-первых, с тем, что наличие четырех α-глобиновых генов способствует образованию адекватного количества α-цепей до тех пор, пока не утрачиваются три или четыре гена. Значительный дисбаланс гемоглобиновых цепей возникает только в том случае, если поражаются три из четырех генов. Во-вторых, агрегаты β-цепей (β1-тетрамеры образуются при недостаточности α-цепей) более растворимы, чем α4-тетрамеры, и поэтому даже у больных с существенно нарушенным синтезом α-глобина при α-талассемии гемолиз гораздо слабее, а эритропоэз более эффективен, чем при β-талассемии.
[17], [18], [19], [20], [21], [22]
Гемоглобинопатия Н — возникает вследствие утраты или дисфункции трех α-глобиновых генов. Клиническая картина такая же как при промежуточной форме β-талассемии. Заболевание проявляется к концу первого года жизни умеренной хронической гемолитической анемией (Нb 80-90 г/л); на фоне интеркуррентных заболеваний или при приеме лекарственных препаратов могут развиваться гемолитические кризы с падением уровня гемоглобина до 40 г/л, требующие гемотрансфузий. Могут отмечаться отставание в физическом развитии, монголоидный тип лица, желтуха, гепатоспленомегалия. В анализах крови — гипохромная анемия, ретикулоцитоз, микроцитоз, анизо- и пойкилоцитоз, мишеневидные эритроциты. Большинство эритроцитов содержит НbН, который представляет собой β4-тетрамеры, образующиеся при избытке β-цепей, и выявляется при электрофорезе в геле как наиболее подвижная фракция. НbН преципитирует в основном в зрелых эритроцитах, что сопровождается умеренной гемолитической анемией. При электрофорезе гемоглобина уровень НbН составляет 5-30 %. Определяется также гемоглобин Барта в различных количествах. Содержание НbА2 снижено, HbF — в нормальных количествах или слегка повышено (до 3 %).
Показания к началу трансфузионной терапии:
- большая форма β-талассемии, гемоглобинопатия Н при уровне гемоглобина ниже 70 г/л;
- промежуточная и большая формы β-талассемии, гемоглобинопатия Н при уровне гемоглобина 70-90 г/л при выраженном отставании физического развития, наличии костных изменений, значительном увеличении селезёнки.
источник
Навигация: Начало Оглавление Поиск по книге Другие книги  — 0
— 0
 — 0
— 0Под талассемией понимают группу наследственных заболеваний, проявляющихся нарушением синтеза какой‑либо из цепей глобина. При данной форме патологии отмечается гипохромная анемия при нормальном либо повышенном содержании железа сыворотки.
Рассматриваемое заболевание впервые было описано американскими педиатрами в 1925 г. Они наблюдали 5 детей из семей итальянцев‑эмигрантов. У детей обнаружили признаки гипохромной анемии тяжелого течения, значительное увеличение печени и селезенки, костные изменения. После опубликования сообщения появилась работа итальянских авторов, описавших сходную, но значительно более легкую форму течения талассемии, при которой лица, страдающие указанной патологией, доживали до зрелого возраста.
Термин «талассемия» был предложен в 1936 г. Болезнь Кули стали называть большой талассемией (thalassemia major ), рассматривая ее как гомозиготную форму наследственной патологии. Впервые мысль о том, что талассемия является результатом нарушения синтеза цепей глобина, высказали независимо несколько ученых. Талассемия, при которой нарушается синтез β‑цепи глобина, называется β‑талассемией; при α‑талассемии нарушается синтез α‑цепи. Описаны также случаи γ‑, δ‑, δ‑талассемии с нарушением синтеза соответствующих цепей глобина. Чаще встречается β‑талассемия.
При талассемии одна из цепей глобина синтезируется в малом количестве или совсем не синтезируется. В норме синтез цепей глобина сбалансирован. Количество α– и не α‑цепей одинаково, и свободных цепей глобина в норме не бывает. Нарушенный синтез одной из них приводит к нарушению баланса. Цепь, которая производится в избыточном количестве, агрегирует и откладывается в эритрокариоцитах. С этим связана большая часть клинических проявлений талассемии.
При α‑талассемии в большинстве случаев происходит потеря участка хромосомы структурных генов, отвечающих за синтез α‑цепи, она кодируется не одной, а у большинства людей двумя парами генов, расположенных в хромосоме 11. Отсутствие α‑цепи у плода приводит к развитию водянки и внутриутробной смерти. Делеция в одном из 4 генов, кодирующих α‑цепь, вызывает легкий дефицит α‑цепи; делеция в 2 генах – более выраженный дефицит, но клинические проявления болезни во многом зависят от того, какие 2 гена не функционируют – в одной хромосоме или в разных. Неодинаково значение 2 пар генов, ответственных за синтез α‑цепи: одна пара главная, другая второстепенная. Клиническая картина зависит и от того, в каких 2 из 4 генов произошла мутация. Если отсутствуют 3 гена, то у больных имеется гемоглобинопатия Н. Гемоглобин Н состоит из 4 β‑цепей. Он очень нестоек, агрегирует, и при этом развивается выраженная гемолитическая анемия.
Значительно более сложен механизм β‑талассемии. Синтез β‑цепи нарушается при ряде различных заболеваний: β‑талассемии, наследственном персистировании фетального гемоглобина, гемоглобинопатии Lepore . Однако при этих заболеваниях нарушения неодинаковы. Ген, кодирующий синтез β‑цепи, располагается в хромосоме 16. В этой же хромосоме рядом с ним располагаются гены, ответственные за синтез γ– и δ‑цепей. При β‑талассемии, когда β‑цепь вообще не производится, не удалось обнаружить делеции гена. При талассемии имеются признаки нестабильности РНК. Основной причиной части случаев β‑талассемии является нарушение сплайсинга (процесс «сшивки» кодирующих участков). Под сплайсингом понимают изменения, которым подвергается мРНК по пути от ядра, где она синтезируется, в цитоплазму. Первичные транскрипты мРНК устроены так, что участки, кодирующие белок, чередуются с участками, его не кодирующими. Эти некодирующие участки в норме вырезаются из молекулы РНК, а кодирующие части молекулы соединяются друг с другом. Нарушение сплайсинга может приводить к нарушению стабильности мРНК.
Делеция гена, ответственного за синтез δ‑цепи, выявлена и при других формах патологии. Это β‑, δ‑талассемия. Делеция гена β– и δ‑цепей обнаружена при наследственном персистировании фетального гемоглобина. Это своеобразное заболевание с очень легкой анемией или без нее, когда содержание фетального гемоглобина сильно увеличено (до 95–98%). В отличие от гомозиготной β‑талассемии, при которой иногда обнаруживается такое же высокое содержание фетального гемоглобина, при наследственном персистировании фетального гемоглобина нет нарушения баланса в синтезе цепей. Гемоглобинопатия Lepore – следствие делеции в генах, ответственных за синтез δ– и γ‑цепей.
В основе клинических проявлений талассемии основное значение придается избыточному количеству цепи глобина. Так, при β‑талассемии в связи с нарушением синтеза β‑цепи оказывается много свободных α‑цепей. Избыточный синтез α‑цепи является главной причиной неэффективного кроветворения при β‑талассемии. Эритрокариоциты погибают в костном мозге. Гибель эритрокариоцитов и в меньшей степени ретикулоцитов и эритроцитов периферической крови в селезенке приводит к выраженному малокровию. При этом в селезенке и в печени могут формироваться очаги красного кроветворения. Интенсивное кроветворение в костях может привести к их искажению, выраженная гипоксия – к нарушению развития ребенка. Существует корреляция между глубиной анемии и избытком α‑цепи при β‑талассемии. При более высоком содержании фетального гемоглобина клетки разрушаются меньше. Это связано с тем, что избыточные α‑цепи находятся не в свободном, а в связанном с γ‑цепями состоянии в виде гемоглобина F. У некоторых больных гомозиготной талассемией нет тяжелых признаков болезни. Анемия у них не столь глубока, они могут жить без постоянных гемотрансфузий, и клинически болезнь определяют не как большая талассемия, а как промежуточная талассемия. Имеются больные более тяжелой гетерозиготной β‑талассемией. У этих больных нередко обнаруживаются включения выпавших в осадок избыточных α‑цепей. Тяжесть гетерозиготной талассемии у этих больных обусловлена недостаточной способностью клеток освобождаться от избыточных α‑цепей. Обычно при гетерозиготной талассемии отмечается компенсаторное повышение утилизации избыточных α‑цепей.
При α‑талассемии в случае отсутствия синтеза α‑цепи и в связи с этим гемоглобинов А, А2 и F развивается водянка плода, которая приводит его к гибели. Избыток β‑цепей при талассемии способен образовывать гемоглобин, состоящий из 4 β‑цепей, – гемоглобин Н. Клетки, содержащие гемоглобин Н, очень легко удаляются из циркуляции селезенкой. Анемия при гемоглобинопатии Н обусловлена как гемолизом периферических эритроцитов, так и нарушением синтеза глобина.
При гомозиготной β‑талассемии, которая описана в 1925 г., клинические проявления отмечаются к концу первого или, реже, второго года жизни ребенка. В первые месяцы жизни обнаруживается лишь умеренная анемия, и далеко не всегда ясно, унаследовал ребенок болезнь от одного или обоих родителей.
Гомозиготная β‑талассемия сопровождается значительным увеличением селезенки, желтушностью и сероватым оттенком кожи и слизистых оболочек, выраженной бледностью. Резкая гиперплазия кроветворного костного мозга приводит к самым разнообразным деформациям скелета: отмечаются практически квадратный череп, сильно сплющенная переносица, сужение глазных щелей.
При рентгенологическом исследовании выявляют увеличение толщины губчатого слоя костей свода черепа, а также поперечную исчерченность на наружной пластинке лобной и теменной костей. Дети отстают в физическом развитии, у них снижена сопротивляемость различным инфекциям, задерживается и даже нарушается половое развитие.
Содержание гемоглобина снижается до 30–50 г/л. Выявляется развитая гипохромия эритроцитов, цветовой показатель, как правило, 0,5 и ниже. Количество ретикулоцитов увеличено, раздражение красного ростка гораздо выраженнее, чем соответствующий этому раздражению ретикулоцитоз. Содержание ретикулоцитов повышается до 2,5–4%. Содержание железа сыворотки чаще всего повышено, но может не выходить за верхнюю границу нормы. В костном мозге содержание сидеробластов увеличено.
Повышение уровня билирубина за счет непрямой фракции является характерной особенностью гомозиготной β‑талассемии.
Вследствие чрезмерного отложения железа формируются цирроз печени, кардиосклероз, сахарный диабет. Также отмечается недоразвитие вторичных половых признаков.
По степени тяжести различают: тяжелую гомозиготную талассемию, при которой больные умирают на первом году жизни, среднетяжелую, при которой дети доживают до 5–8 лет, и более легкую форму, позволяющую больным дожить до взрослого возраста.
Диагноз гомозиготной β‑талассемии подтверждается исследованием содержания фетального гемоглобина в эритроцитах. При гомозиготной талассемии данный показатель увеличивается до 20–30.
Гетерозиготная β‑талассемия – результат наследования болезни только от одного из родителей, возможно, с бессимптомным течением либо с нерезкими клиническими проявлениями. Наиболее редко встречаются бурные клинические проявления. Жалобы больных гетерозиготной талассемией носят общий характер и проявляются слабостью, повышенной утомляемостью, понижением работоспособности, наиболее тяжело больной воспринимает физический труд. Кожные покровы у таких больных бледные, характерна небольшая иктеричность кожи и склер.
Увеличение селезенки – достаточно часто встречаемый симптом гетерозиготной β‑талассемии.
Показатели крови при гетерозиготной талассемии крайне разнообразны. Содержание гемоглобина может быть близко к норме. Так называемая thalassemia minima сопровождается показателями гемоглобина 110–120 г/л, либо он снижен до 90–100 г/л (thalassemia minor ), редко показатель достигает 70 г/л. Анемия носит гипохромный характер. Снижение цветового показателя может быть значительным (0,5–0,7). Кроме гипохромии эритроцитов, часто выявляют анизоцитоз и пойкилоцитоз, мишеневидность эритроцитов.
Также мишеневидность эритроцитов обнаруживается при железодефицитной анемии, свинцовой интоксикации, у лиц, перенесших спленэктомию.
Типичным признаком талассемии служат базофильно пунктированные эритроциты.
Содержание ретикулоцитов при гетерозиготной талассемии обычно повышается до 2–5%. Обнаруживается значительное раздражение красного ростка костного мозга. Нередко красный росток в 1–3 раза превышает белый. Уменьшено количество зрелых эритрокариоцитов, содержащих гемоглобин. Содержание гранул железа в костном мозге увеличено или, реже, нормально. Содержание железа сыворотки у больных гетерозиготной талассемией нормальное, реже повышенное. Запасы железа, определенные по десфераловому тесту, оказываются повышенными.
В 75% случаев гетерозиготной β‑талассемией выявляется повышение уровня непрямого билирубина.
Диагноз гетерозиготной β‑талассемии ставится на основании повышения содержания фракции гемоглобина А2. Содержание гемоглобина А2 при гетерозиготной β‑талассемии увеличивается до 4,2–8,9% общего количества гемоглобина. Приблизительно у половины больных обнаруживается увеличение содержания фетального гемоглобина до 2,5–7%.
Важным диагностическим признаком является подобное заболевание у членов семьи больного.
Синтез α‑цепи глобина в отличие от синтеза β‑цепи контролируется двумя парами неодинаковых по значению генов, в связи с этим существуют различные варианты течения α‑талассемии.
При полном отсутствии α‑цепи, гомозиготном нарушении функции всех 4 генов у плода не синтезируется фетальный гемоглобин, развивается водянка, в результате чего наступает смерть. Нарушение функции 1 или 2 генов приводит к анемии, как правило, нетяжелого течения. Клинические проявления α‑талассемии с поражением 2 генов зависят от того, какие гены поражены, и почти полностью повторяют гетерозиготную β‑талассемию. Нередко обнаруживается увеличение селезенки, реже – печени.
Определяются умеренная гипохромная анемия с мишеневидными эритроцитами и эритроцитами с базофильной пунктацией, незначительное повышение уровня ретикулоцитов, небольшая гипербилирубинемия и повышение железа сыворотки, повышение осмотической резистентности эритроцитов. Отмечается резкое раздражение красного ростка костного мозга. Как правило, кровные родственники больного имеют такую же анемию.
Однако в отличие от β‑талассемии при α‑талассемии не увеличивается количество фетального гемоглобина и гемоглобина А2. α‑талассемию удается диагностировать только в случае изучения биосинтеза цепей глобина in vitro (в пробирке).
Альфа‑талассемию иногда выявляют у новорожденных: при исследовании крови обнаруживается гемоглобин Bart , лишенный α‑цепи. Этот гемоглобин состоит из 4 γ‑цепей.
Гемоглобинопатия Н является одним из вариантов α‑талассемии. Она сравнительно нетяжелая, проявляется увеличением размеров как селезенки, так и печени, незначительной желтухой, в основном за счет увеличения фракции непрямого билирубина, анемией различной степени выраженности, обычно содержание гемоглобина не опускается ниже 70–80 г/л. Отмечаются выраженная гипохромия эритроцитов, их мишеневидность, базофильная пунктация. Так же как и при других формах талассемии, при гемоглобинопатии Н обнаруживаются признаки неэффективного кроветворения: резкое раздражение красного ростка костного мозга при небольшом повышении уровня ретикулоцитов. Гемоглобинопатия Н отличается от других форм талассемии множественными мелкими включениями во всех эритроцитах.
Гемоглобинопатия Н впервые была описана в 1955 г. Rigas с соавторами. Данные ученые наблюдали 2 больных с гемолитической анемией, у которых при электрофорезе гемоглобина в щелочном буфере была обнаружена дополнительная фракция, движущаяся впереди гемоглобина А.
Гемоглобинопатия Lepore – это формы талассемии, при которых синтез α‑цепи глобина нормальный, а синтез второй цепи нарушен. Вместо нормальной β‑цепи синтезируется своеобразная цепь, состоящая из фрагментов β– и δ‑цепей. Клинические признаки гемоглобинопатии Lepore крайне похожи на таковые при гомозиготной β‑талассемии.
Выявляется около 70–80% фетального гемоглобина и около 20% гемоглобина Lepore . Гетерозиготную форму гемоглобинопатии Lepore по клиническим и гематологическим признакам невозможно отличить от гетерозиготной талассемии. Содержание гемоглобина колеблется в пределах от 90 до 100 г/л, нередко увеличена селезенка. У гетерозигот содержание гемоглобина Lepore составляет 7–8%, при этом снижается содержание гемоглобина А2 и повышается содержание фетального гемоглобина.
Гемоглобинопатии Lepore встречаются редко.
Лечение гомозиготной β‑талассемии . Так как проявления болезни определяются гипоксией и активным кроветворением в костях, где обычно оно отсутствует, проводят переливания эритроцитов больному с раннего возраста.
Вначале применяется ударный курс лечения (за 2–3 недели 8–10 переливаний). При этом содержание гемоглобина обычно повышается до 120–140 г/л. Затем переливания делают реже, каждые 3–4 недели, из расчета 20 мл/кг. Уровень гемоглобина стараются поддерживать на 90–100 г/л. Массивные переливания при талассемии не только улучшают общее состояние, но и уменьшают изменения скелета, увеличение селезенки, улучшают развитие детей, снижают частоту тяжелых инфекций.
Частыми осложнениями данной терапии являются пирогенные реакции, в основном при применении цельной крови или недостаточно отмытых эритроцитов.
Одним из серьезных осложнений переливания является избыточное отложение железосодержащего пигмента, что приводит к существенному увеличению печени, сахарному диабету, сердечной недостаточности. Лечение гомозиготной талассемии включает обязательное использование десферала для выведения избыточного количества железа. Доза десферала (вводится внутримышечно) зависит от количества перелитых эритроцитов и возраста больного: для маленьких детей – 10 мг/кг, для подростков – 500 мг/сут. Допустимы и большие дозы – 1 г/сут. Рекомендуется сочетать десферал с 200–500 мг аскорбиновой кислоты внутрь.
Значительное увеличение селезенки, присоединение лейкопении, тромбоцитопении к признакам гипохромной анемии заставляют думать о спленэктомии.
Лечение гомозиготной талассемии указанными методами улучшает общее состояние больных, увеличивает продолжительность их жизни.
В настоящее время широко применяется пересадка костного мозга больным талассемией, в результате чего в большинстве случаев достигается стойкое улучшение.
Лечения гетерозиготной β‑талассемии в большинстве случаев не требуется. Больные чувствуют себя удовлетворительно. Содержание гемоглобина колеблется в пределах от 90 до 110 г/л. При гетерозиготной β‑талассемии переливаний крови не требуется. Удаление селезенки производится крайне редко и лишь при очень большой селезенке.
Десферал следует применять при гетерозиготной β‑талассемии, когда наблюдается высокое количество сывороточного железа и железа в моче после введения 500 мг десферала. Однако десферал лишь предотвращает сидероз (заболевание человека, вызываемое осаждением в легких пыли, содержащей железо), не приводя к повышению уровня гемоглобина.
При снижении уровня гемоглобина в связи с инфекционными заболеваниями, при беременности необходимо использовать фолиевую кислоту по 0,005 г 1–2 раза в день, так как при неэффективном кроветворении, связанном с талассемией, потребляется значительное количество фолиевой кислоты.
При гетерозиготной талассемии противопоказаны препараты железа, так как всегда есть некоторый избыток железа без клинических проявлений гемосидероза. Однако больные, принимающие препараты железа, чувствуют себя значительно хуже. Наиболее опасно при талассемии парентеральное введение препаратов железа: больные, находившиеся в удовлетворительном состоянии, могут умереть от тяжелого гемосидероза, в частности от недостаточности кровообращения, связанной с сидерозом миокарда.
Лечение α‑талассемии практически не отличается от лечения гетерозиготной талассемии, за исключением гемоглобинопатии Н.
При гемоглобинопатии Н в связи с нестабильностью этого гемоглобина и выпадением его в осадок имеются признаки не только неэффективного кроветворения, но и выраженного разрушения эритроцитов периферической крови. Это разрушение идет преимущественно в селезенке, в результате чего она увеличивается. Основным методом лечения гемоглобинопатии Н при выраженной анемии является удаление селезенки.
источник